Index
- Overview
- Training an organism
specific GlimmerHMM
- Need of a specific
organism training
- Data collection
- Training GlimmerHMM
- Changing the
parameters obtained during training
- Running GlimmerHMM
- Obtaining GlimmerHMM
I. Overrview
GlimmerHMM is a new gene
finder based on a Generalized Hidden Markov Model (GHMM). Although the
gene finder conforms to the overall mathematical framework of a GHMM,
additionally it incorporates splice site models adapted from the GeneSplicer
program.
JIGSAW, GeneZilla, and GlimmerHMM : puzzling
out the features o
The
variable-length
feature states (e.g., exons, introns, intergenic regions) are
implemented using Nth-order interpolated Markov
models (IMM) as described in Delcher et al.
1999 for N=8. Currently, GlimmerHMM's
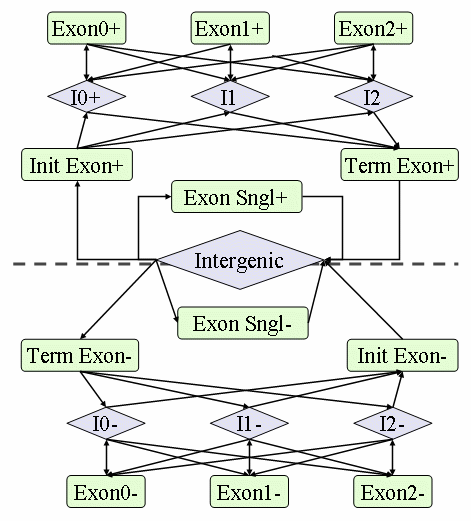
GHMM structure includes introns of each phase, intergenic regions, and
four types of exons (initial, internal, final, and single), as depicted
in the figure below.
There are
a number of assumptions used by GlimmerHMM when predicting genes in the
DNA sequence.
The main assumptions are
(1) the coding region of every gene begins with a
start codon ATG (but partial genes can be predicted),
(2) a gene has no in-frame stop codons except the
very last codon,
(3) each exon is in a consistent reading frame
with the previous exon.
These constraints significantly enhance the
efficiency of computing the optimal gene models, by restricting the
search space of the GHMM algorithm. On the other hand, genuine frame
shifts cannot be detected by the system.
III.Training an organism specific GlimmerHMM
III.1. Need of a specific organism training
Our earlier implementation of a gene finder -
GlimmerM - trained for rice produced clearly
superior gene models than GlimmerM trained for Arabidopsis (Pertea and Salzberg 2002). This is also valid for
GlimmerHMM. Although the current
trainings of GlimmerHMM should work well on closely related organisms,
the user should use our training procedure to re-train the system for
an other eukaryotic organism in order to improve the accuracy of the
gene prediction. For all species that GlimmerHMM is already trained, we
expect the performance to improve even further by re-training the
system as the amount of DNA sequences from these species continues to
grow.
III.2 Data collection
First of all it should be noted that a thorough
collection of a good training data set is a step that should precede
the training of any gene finder. This step is very important since the
quality of the data presented for training is directly proportional
with the accuracy of the resulted gene finder. As any species-specific
gene finder, GlimmerHMM needs a training data set containing as many as
possible complete coding sequences from the organism genome for which
the gene prediction is intended. By surveying the public databases, one
should be able to find all previously known genes of the target
organism that are backed by laboratory evidence. These genes would form
a sound training data set if their number would be large enough.
Unfortunately, this is rarely the case, so one should use other methods
in order to construct a reliable data set. From our experience, most
often the training data set was formed by searching long ORFs (with
more than 500 bases) against non redundant protein sequence databases
by using programs like BLAST in order to map known proteins into the
genome.
III.3 Training GlimmerHMM
To train GlimmerM you should run trainGlimmerM
with the following command:
trainGlimmerHMM <mfasta_file>
<exon_file>
[optional_parameters]
<mfasta_file>
and <exon_file> are
the
multi-FASTA file and the file containing the exon coordinates of the
known genes, respectively.
<mfasta_file>
is a multifasta file containing the sequences for training with the
usual format:
>seq1
AGTCGTCGCTAGCTAGCTAGCATCGAGTCTTTTCGATCGAGGACTAGACTT
CTAGCTAGCTAGCATAGCATACGAGCATATCGGTCATGAGACTGATTGGGC
>seq2
TTTAGCTAGCTAGCATAGCATACGAGCATATCGGTAGACTGATTGGGTTTA
TGCGTTA
<exon_file> is
a file with the exon coordinates relative to the sequences contained in
the <mfasta_file>;
different genes are separated by a blank line; I am assuming a format
like below:
seq1 5 15
seq1 20 34
seq1 50 48
seq1 45 36
seq2 17 20
In this example seq1 has two
genes: one on the direct strand and another one on the complementary
strand. Here
you can find a real example of fasta and exon files.
If not enough data is available for training the
splice sites the training procedure will be unsuccessful and exit with
a warning message. In this case the user should collect more known
genes with introns and then try the training procedure again. If not
enough data is available for training the translational start and stop
sites of
the genes, the user will need to collect more genes. A minimum number
of 50 genes with standard start codons (ATG) and stop codons
(TAA/TAG/TGA) is assumed by default.
The optional parameters that can be given to the
training program are:
-i i1,i2,...,in
|
isochores to be considered for
training (e.g. if two isochores are desired with 0-40% GC content and
40-100% then the option should be: -i 0,40,100; default is set to -i
0,100 )
|
-f val
|
val = average value of upstream UTR
region if known
|
-l val
|
val = average value of
downstream UTR region if known
|
-n val
|
val = average value of
intergenic region if known
|
-v val
|
val = value of flanking region
to be considered around genes (default=200)
|
-b val
|
val = build 1st or 2nd order
markov model for the splice sites (default=1)
|
III.4 Changing the parameters obtained during training
The effects of specific feature states on the
modeling of human
This step is optional to the user but many times a
manual tuning of the system could improve the accuracy of the
predictions. This is due partially to the fact that some of the
parameters required for the training of the system might not be known
for a certain organism (e.g. average size of the intergenic regions) so
one might want to try different values for these
parameters. We noticed that a gradient-ascent procedure which search
through the parameter space similar to the one described in Majoros and Salzberg 2004 will often optimize
the accuracy of the training.
Choosing adequate thresholds that determine the false negative-false
positive ratio for specific site detection may be a challenging problem
given the small number of true sites and the overwhelming number of
false positives, and requires a suite of decisions that maximizes the
accuracy of the recognition task without a big loss in the sensitivity.
In the training procedure of GlimmerHMM the threshold for calling a
sequence a real splice site is chosen by examining the trade-off in the
false positive rate.
The system creates a sorted list of thresholds, adjusting the scoring
function so that it will miss 1,2,3, etc. true sites. The default
threshold is chosen to be the score at which the false positive rate
drops by less than 1%. To allow a greater flexibility in setting the
signals' thresholds, our training procedure allows the user to consult
the false positive and false negative rates specific for the training
data and set his own threshold.
After running trainGlimmerHMM, a log file can be
consulted to find the default values set for some of the parameters of
GlimmerHMM. The file config.file
in the training directory specifies for each isochore which
configuration file to use. For the default case, when no isochores are
considered, there will be only one line with the foillowing info:
C+G <= 100 train_0_100.cfg
All changes in parameters can be done in the train_0_100.cfg file. Each
parameter appears on a single line of the .cfg file. The parameters that can
be changed are described in the following table:
line in the .cfg file
|
Description
|
acceptor_threshold
value
|
acceptor site threshold value;
the false negative/false positive rates for different acceptor
thresholds can be consulted from the false.acc
file.
|
donor_threshold
value
|
donor site threshold value;
the false negative/false positive rates for different donor thresholds
can be consulted from the false.don
file. |
ATG_threshold value
|
start codon threshold value;
the false negative/false positive rates for different start codon
thresholds can be consulted from the false.atg
file. |
Stop_threshold value
|
stop codon threshold value;
the false negative/false positive rates for different stop codon
thresholds can be consulted from the false.stop
file. |
split_penalty value
|
a factor penalizing the gene
finder's tendency to split genes.
|
intergenic_val value
|
the minimum intergenic
distance espected between genes.
|
intergenic_penalty value
|
a penalty factor for genes
situated closer than the intergenic_val value.
|
MeanIntergen value
|
the average size of intergenic
regions.
|
BoostExon value
|
a factor to increase the
sensitivity of exon prediction.
|
BoostSplice value
|
a factor to increase the score
of good scoring splice sites.
|
BoostSgl value
|
a factor to
increase the predicted number of single exon genes.
|
onlytga value
|
set this value
to 1 if TGA is the only stop codon in the genome.
|
onlytaa value
|
set this value
to 1 if TAA is the only stop codons in the genome. |
onlytag value
|
set this value
to 1 if TAG is the only stop codons in the genome. |
IV. Running GlimmerHMM
The program GlimmerHMM takes two inputs: a DNA
sequence file in FASTA format and a directory containing the training
files for the program. If not specified, the training directory is by
default the current working directory. No options are implemented
at this time for the program.
VI. Obtaining GlimmerHMM
GlimmerHMM is available free of charge under the
open-source Artistic
License.

To download GlimmerHMM please click here.
References
Delcher, A.L., Harmon, D., Kasif, S., White,
O., and Salzberg, S.L. Improved microbial
gene identification with GLIMMER Nucleic Acids Research,
27:23 (1999), 4636-4641.
Majoros, W.H., Pertea, M.,
and Salzberg, S.L.
TigrScan
and GlimmerHMM: two open-source ab initio eukaryotic gene-finders Bioinformatics 20 2878-2879.
Majoros,W.M. and Salzberg, S.L. (2004) An
empirical analysis of training protocols for probabilistic gene
finders. BMC Bioinformatics 5:206.
Pertea, M., X. Lin, et al. (2001). "GeneSplicer: a
new computational method for splice site prediction." Nucleic Acids Res
29(5): 1185-90.
Pertea, M. and S. L. Salzberg (2002).
"Computational gene finding in plants." Plant Molecular Biology
48(1-2): 39-48.
Pertea, M., S. L. Salzberg, et al. (2000).
"Finding genes in Plasmodium falciparum." Nature 404(6773): 34;
discussion 34-5.
Salzberg, S. L., M. Pertea, et al. (1999).
"Interpolated Markov models for eukaryotic gene finding." Genomics
59(1): 24-31.
Yuan, Q., J. Quackenbush, et al. (2001). "Rice
bioinformatics. analysis of rice sequence data and leveraging the data
to other plant species." Plant Physiol 125(3): 1166-74.
Back to the CBCB Software
Page
|