CBCB Seminar Series
Most CBCB seminars are held from 2
p.m. until 3:15 p.m. on Thursdays
in the CBCB seminar room, 3118 at Biomolecular Sciences Building #296
Some external seminars are listed here. These, and some other
exceptions, will have a different time and/or place.
For directions to CBCB please scroll down.
2:00 pm Thursday, Feb. 11, 2010
Title: " HMMER: a new generation of homology search software"
By: Sean Eddy
Venue: 1103 Biosciences Research Bldg.
Abstract:Database homology searching might be the most important application in computational molecular biology, and since the 1990s, BLAST has been our main workhorse. Since BLAST's introduction, theoretical advances have been made in applying probabilistic inference methods to homology searches using hidden Markov model (HMM) approaches. General adoption of probabilistic methods has been limited by some key problems, including the fact that the popular HMM implementations (including my HMMER software) are computationally demanding. I will talk about HMMER3, a new generation of HMMER that aims to even more fully deploy probabilistic inference technology on homology searches, while at the same time attaining (and perhaps soon surpassing) BLAST's speed.
Speaker information:Sean Eddy is a group leader at Janelia Farm.
This talk was canceled due to snow and has been rescheduled for Sept. 8, 2011.
11:30 a.m. Monday, Feb. 22, 2010
Title: "Copy number
variation detection from SNP genotyping data and next-generation
sequencing data"
By: Kai Wang
Venue: 1140 BPS Biology-Psychology
Speaker information:
kai.genotypic.com
2:00 p.m. Wednesday, Feb. 24, 2010
Title: "Protein interaction
networks in viruses and bacteria"
By: Peter Uetz
Venue: 1103 BRB
Speaker information: JCVI web site
3:00-4:30 p.m. Wednesday, March 3, 2010
Title: "A Teaching Career to Facilitate Student Learning"
By: Malcolm Campbell
Venue: 1103 BRB (Bioscience
Research Bldg., not Biomolecular Sciences)
More information: Dr. Malcolm
Campbell is a biology professor at Davidson College, NC and the
founding director
of the Genome Consortium for Active Teaching (GCAT). This
presentation is hosted by the Teaching
and Learning Center of the College of Chemical and Life Sciences.
2:00 p.m., Thursday, March 4, 2010
Title: "Undergraduates Use Synthetic Biology to Build Bacterial Computers"
By: Malcolm
Campbell
Venue: 3118 Biomolecular
Sciences
Week of March 8. Special Seminars by Prof. Zhaobang Zeng, NC State University
Date and Time: Monday, March 8, 4:00 pm
Title: "Mapping multiple QTLs with epistasis: theory, method and practice"
Date and Time: Tuesday, March 9, 4:00 pm
Title: "eQTL Mapping analysis"
Date and Time: Wednesday, March 10, 4:00 pm
Title: "Identifying network structure from eQTL mapping and Epistasis and Gene pathway inference"
Date and Time: Thursday, March 11, 2:00
Title: "Study genetic basis and pathways of complex traits"
By: Dr. Zhao-Bang Zeng, North Carolina State University
Venue: 0467 Animal Sciences (reading room)
Speaker information: Zhao-Bang Zeng
Host: Jiuzhou Song Ph.D, Animal Sciences
2:00 p.m. Thursday, March 25, 2010
Title: Computational discovery of epistatic interactions in P. falciparum - A tale of two loci
By: Yang Huang,
Computational Biology Branch, National Center for
Biotechnology Information, National Library of Medicine, NIH
Venue: 3118 Biomolecular Sciences (as usual)
Abstact:
Identification of epistatic interactions between genomic loci is fundamental
for understanding genome organization and gene regulation. In the last decade,
expression Quantitative Trait Loci (eQTL) studies have been widely used to
determine the relation between single-locus genotype and gene expression.
However, computational and statistical challenges have limited genome-wide
studies of epistatic interactions affecting gene expression. Previously, we
developed a Graph based eQTL Decomposition method (GeD) that allows us to model
genotype and expression data using an eQTL association graph. Based on the eQTL
association graph, We developed a new method for reliable detection of
epistatic interactions that can overcome some of the statistical limitations of
classical methods and applied it to uncover epistatic interactions among
genomic loci of the human malaria parasite P. falciparum. In the landscape of
epistatic interactions of this parasite segregating in a set of 34 haploid
progeny, we found numerous hotspots with potentially important regulatory
functions. We also observed that elevated linkage disequilibrium (LD) between
two loci on different chromosomes in P. falciparum correlates with the number
of regulated target genes regulated jointly by these loci. Such results
indicate LD's important role in maintaining parasite specific, biological
functions.
2:00 p.m., Thursday, April 8, 2010
Title: "From genomics data
integration to using functional relationship networks to understand
disease at the molecular level"
By: Olga Troyanskaya
Venue: 3118 Biomolecular Sciences
(directions)
Speaker information: Olga Troyanskaya
2:00 p.m., Friday, April 9, 2010
Title: "Transcript Assembly and Abundance Estimation with High-Throughput RNA Sequencing"
By: Cole Trapnell,
CBCB
Venue: 3118 in CBCB (our usual venue)
Abstact:
We present algorithms and statistical methods for the
reconstruction and abundance estimation of transcript sequences from high
throughput RNA sequencing ("RNA-Seq"). We evaluate these approaches through
large-scale experiments of a well-studied model of muscle development.
We begin with an overview of sequencing assays and outline why the short
read alignment problem is fundamental to the analysis of these assays. We
then describe two approaches to the contiguous alignment problem, one of
which uses massively parallel graphics hardware to accelerate alignment, and
one of which exploits an indexing scheme based on the Burrows-Wheeler
transform. We then turn to the spliced alignment problem, which is
fundamental to RNA-Seq, and present an algorithm, TopHat. TopHat is the
first algorithm that can align the reads from a large RNA-Seq experiment to
a mammalian-sized genome without the aid of reference gene models.
In the second part of the thesis, we present the first comparative RNA-Seq
assembly algorithm, Cufflinks, which is adapted from a constructive proof of
Dilworth'sTheorem, a classic result in combinatorics. We evaluate Cufflinks
by assembling the transcriptome from a time course RNA-Seq experiment of
developing skeletal muscle cells. The assembly contains 13,689 known
transcripts and 3,724 novel ones. Of the novel transcripts, 62% were
strongly supported by earlier sequencing experiments or by homologous
transcripts in other organisms. We further validated interesting genes with
isoform-specific RT-PCR.
We then present a statistical model for RNA-Seq included in Cufflinks and
with which we estimate abundances of transcripts from RNA-seq data.
Simulation studies demonstrate that the model is highly accurate. We apply
this model to the muscle data, and track the abundances of individual
isoforms over development.
Finally, we present significance tests for changes in relative and absolute
abundances between time points, which we employ to uncover differential
expression and differential regulation. By testing for relative abundance
changes within and between transcripts sharing a transcription start site,
we find significant shifts in the rates of alternative splicing and promoter
preference in hundreds of genes, including those believed to regulate muscle
development.
A Dissertation Defense for the degree of Ph.D. in Computer Science
2:00 p.m., Wednesday, April 14, 2010
Title: "Whole-Genome Sequence Analysis for Pathogen Detection and Diagnostics"
By: Adam Phillippy,
CBCB
Venue: 3118 Biomolecular Sciences
Abstact:
Pathogenic microbes, both natural and weaponized, pose
significant dangers to human health and safety. To defend against these
threats, it is essential to rapidly detect and characterize pathogens in any
environmental or clinical medium with high accuracy. Now that the genome
sequences of thousands of bacteria and viruses are known, it is possible to
design biomolecular tests to rapidly detect and characterize pathogens based
solely on their DNA. Possible applications are far-reaching and include
real-time clinical diagnosis and biosurveillance. However, these tests
require sophisticated computational design and analysis to operate
effectively.
This dissertation presents novel computational methods for improving the
accuracy of three modern diagnostic technologies: polymerase chain reaction
(PCR), array comparative genomic hybridization (CGH), and whole-genome
sequencing. For designing real-time PCR detection assays, an efficient
search algorithm and data structure are presented for analyzing over 100
billion nucleotides of genomic DNA to identify the most distinguishing
sequences of a pathogen. Laboratory validation shows that these "signature"
sequences can be used to detect pathogens in complex samples and
differentiate them from their non-pathogenic relatives. For CGH, pan-genome
array design and analysis algorithms are presented for the characterization
of microbial isolates. These methods are used to study multiple strains of
the foodborne pathogen, Listeria monocytogenes, revealing new insights into
the diversity and evolution of the species. Finally, multiple methods are
presented for the validation of whole-genome sequence assemblies. These
validated assemblies provide the ultimate diagnostic, decoding the entire
DNA sequence of a genome with high confidence.
A Dissertation Defense for the degree of Ph.D. in Computer Science
2:00 p.m., Thursday, April 15, 2010
RECOMB 2010 Practice Talks
Venue: 3118 Biomolecular
Sciences
directions
Title (RECOMB 2010 Practice
Talk): "Dense Subgraphs with
Restrictions and Applications to Gene Annotation Graphs"
Authors: Barna Saha, Allison
Hoch, Samir Khuller, Louiqa Raschid and Xiao-Ning Zhang
Speaker: Barna Saha,
a third year Computer Science graduate student working with Samir
Khuller on
algorithm
design and analysis.
Abstract:
We focus on finding complex annotation patterns representing
novel and interesting hypotheses from gene annotation data.
We define a generalization of the densest subgraph problem by adding
an additional distance restriction (defined by a separate metric)
to the nodes of the subgraph.
We show that while this generalization makes the problem NP-hard
for arbitrary metrics,
when the metric comes from the distance metric of a tree, or an
interval graph, the problem can be solved optimally in polynomial time.
We also show that the densest subgraph problem with a specified
subset of vertices that have to be included in the solution
can be solved optimally in polynomial time. In addition, we consider
other extensions when not just one solution needs to be found, but
we wish to list all subgraphs of almost maximum density as well.
We apply this method to a dataset of genes and their annotations
obtained from The Arabidopsis Information Resource (TAIR).
A user evaluation confirms that the patterns found in the distance
restricted densest subgraph for a dataset of photomorphogenesis genes
are indeed validated in the literature; a control dataset validates
that these are not random patterns. Interestingly, the complex
annotation patterns potentially lead to new and as yet unknown
hypotheses.
We perform experiments to determine the properties of the dense
subgraphs, as we vary parameters, including the number of genes and the
distance.
-------------
Title (RECOMB 2010 Practice
Talk): "Extracting between-pathway models from E-MAP interactions using expected graph compression"
Speaker: David Kelley
Abstract:
Genetic interactions (such as synthetic lethal interactions)
have become quantifiable on a large-scale using the epistatic miniarray
profile (E-MAP) method. An E-MAP allows the construction of a large,
weighted network of both aggravating and alleviating genetic interactions
between genes. By clustering genes into modules and establishing relationships
between those modules, we can discover compensatory pathways.
We introduce a general framework for applying greedy clustering
heuristics to probabilistic graphs.We use this framework to apply a graph
clustering method called graph summarization to an E-MAP that targets
yeast chromosome biology. This results in a new method for clustering
E-MAP data that we call Expected Graph Compression (EGC). We validate
modules and compensatory pathways using enriched Gene Ontology
annotations and a novel method based on correlated gene expression
from a comprehensive collection of expression experiments. EGC finds a
number of modules that are not found by any of the previous methods to
cluster E-MAP data. Further, EGC uncovers core submodules contained
within several previously found modules, suggesting that EGC can reveal
the finer structure of E-MAP networks.
1:00 p.m., Friday, April 16, 2010
Title: " High Performance Computing for DNA Sequence Alignment and Assembly"
By: Michael C. Schatz,
CBCB
Venue: 3118 Biomolecular Sciences
Abstact:
We are at the dawn of a new era in computational biology. DNA
sequencing projects that required years of effort and hundreds of millions
of dollars of equipment just a few years ago, can now be performed quickly
and cheaply by individual labs. This dramatic shift is expanding the scale
and scope of sequencing to previously unimaginable limits, and will
ultimately lead to new discoveries about our basic biology, the diversity of
life, and personalized medicine. However, these ambitious goals can only be
realized if we can develop new computational methods that can effectively
analyze the overwhelming volumes of data generated.
In my presentation, I'll describe my research developing efficient methods
for analyzing large biological datasets, including by using highly parallel
commodity graphics processing units produced by nVidia, and the parallel
computing framework MapReduce developed by Google. My dissertation research
demonstrates how these technologies can be applied to the critical tasks of
large-scale alignment and assembly, enabling genotyping and de novo assembly
of whole genome genomes from billions of short reads. Coupled with
inexpensive cloud computing, these programs can quickly, cheaply, and
accurately analyze tremendous biological datasets and have the potential to
make otherwise infeasible studies practical.
A Dissertation Defense for the degree of Ph.D. in Computer Science
2:00 p.m., Thursday, April 29, 2010
Title:
"Structural Assembly of Molecular Complexes Based on Residual Dipolar Couplings"
Speaker: Konstantin
Berlin, a finishing PhD student in Computer Science
Venue: 3118 Biomolecular
Sciences
directions
Abstact:
We present PATI, a computationally efficient and accurate abinitio predictor of the residual dipolar couplings (RDCs) from a protein structure. Building upon PATI, we develop and evaluate a rigid-body molecular docking method, called PATIDOCK, that relies solely on the three-dimensional structure of the individual components and the experimentally derived RDCs for the complex, and show that it is possible to accurately assemble a protein-protein complex by utilizing PATI to guide the docking method. The proposed docking method is robust against experimental errors in the RDCs and computationally efficient. We analyze the accuracy and efficiency of this method using experimental or synthetic RDC data for several proteins, as well as synthetic data for a large variety of protein-protein complexes. We also test our method on two protein systems for which the structure of the complex and steric-alignment data are available (Lys48-linked diubiquitin and a complex of ubiquitin and a ubiquitin-associated domain) and analyze the effect of flexible unstructured tails on the outcome of docking. The results demonstrate that it is fundamentally possible to assemble a protein-protein complex based solely on experimental RDC data and the prediction of the alignment tensor from three-dimensional structures. Additionally we show a method for combining RDCs with other experimental data, such as ambiguous constraints from interface mapping, to further improve structure characterization of the protein complexes.
Past Events
Other Events
- UMCP
Bioscience Research & Technology Review Day (November 12, 2009)
- UMCP
Bioscience Research & Technology Review Day (November 12, 2008)
- UMCP
Bioscience Research & Technology Review Day (November 13, 2007)
- UMCP
Bioscience Research & Technology Review Day (November 16, 2006)
- UMCP
Bioscience Research & Technology Review Day (November 17, 2005)
- UMCP Hosts NIH Bioinformatics Course (January 19 & 20, 2005)
- UMCP
Bioscience Research & Technology Review Day (November 4, 2004)
- UMCP Hosts NIH Bioinformatics Course (January 22 & 23, 2004)
- UMCP
Bioscience Research & Technology Review Day (November 5, 2003)
- UMCP Hosts NIH Bioinformatics Course (January 15 & 16, 2003)
Directions
More detailed transportation options to CBCB can be found here.
From the Capital Beltway to Parking
Lot:
- take Capital Beltway (I-495) Exit 25 and turn onto Baltimore
Avenue (US Route 1) South
- go two miles south on Baltimore Ave and enter the main gate at
Campus Drive
- take the right lane into campus and make first right turn onto
Paint Branch Drive
- stop at the first stop sign then pass Stadium Drive on the left
- stop at the second stop sign then pass Parking Lot XX2 on the
right
- look for the Paint Branch Drive Visitor Lot on the left
- turn left onto Technology Drive and park in the Paint Branch
Drive Visitor Lot
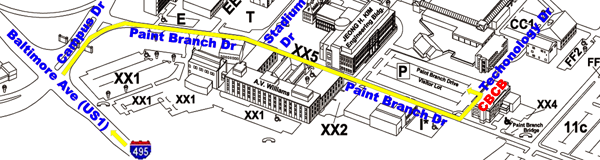
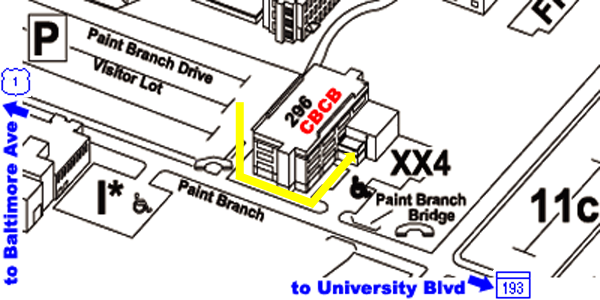
From Parking Lot to CBCB:
- the back of the Biomolecular Sciences Building #296 faces this
parking lot
- walk around to the front of the building and using the keypad
near the front door
- dial the number of one of the CBCB staff members in order to
gain entrance to the building
- CBCB is located on the third floor of the Biomolecular Sciences
Building #296
|
|
