What is TWAIN?
TWAIN is a new syntenic genefinder which employs a Generalized Pair
Hidden Markov Model (GPHMM) to predict genes in two closely related
eukaryotic genomes simultaneously. It utilizes the MUMmer
package to perform approximate alignment before applying a GPHMM based
on an enhanced version of the TigrScan
gene finder. TWAIN was written by Bill Majoros and Mihaela Pertea while at The Institute for
Genomic Research (TIGR).
TWAIN is available for download as open source
software.
Results on Aspergillus
fumigatus vs. A. nidulans:
|
Nuc
Sens
|
Nuc
Spec
|
Nuc
Accur
|
Splice
Sens
|
Splice
Spec
|
Start/
Stop
Sens
|
Start/
Stop
Spec
|
Exon
Sens
|
Exon
Spec
|
Exact
Genes
|
Genscan
(no homology)
|
96%
|
99%
|
95%
|
66%
|
69%
|
57%
|
58%
|
50%
|
52%
|
22%
|
TigrScan
(no homology)
|
99% |
100% |
99% |
89% |
81% |
81% |
80% |
78% |
73% |
54% |
TWAIN
(using homology)
|
99% |
100% |
99% |
94% |
88% |
92% |
92% |
89% |
85% |
74% |
(N=147 gene pairs)
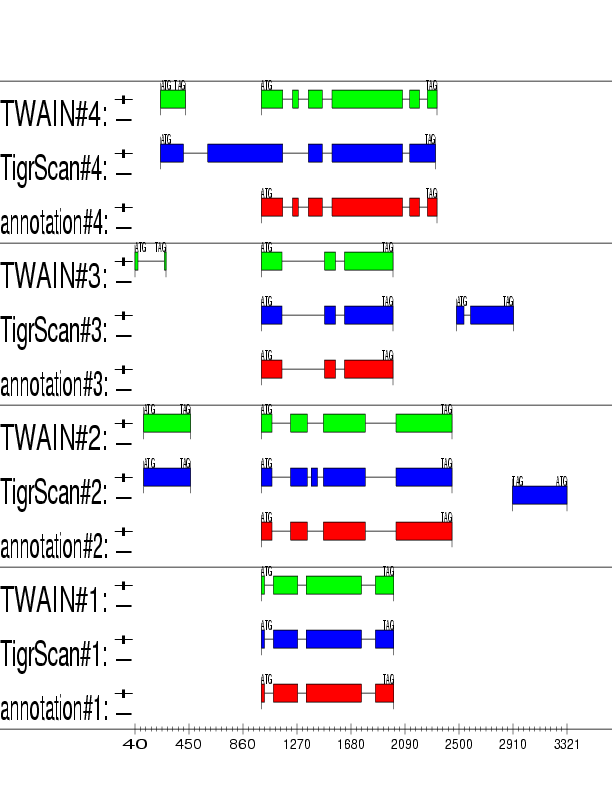
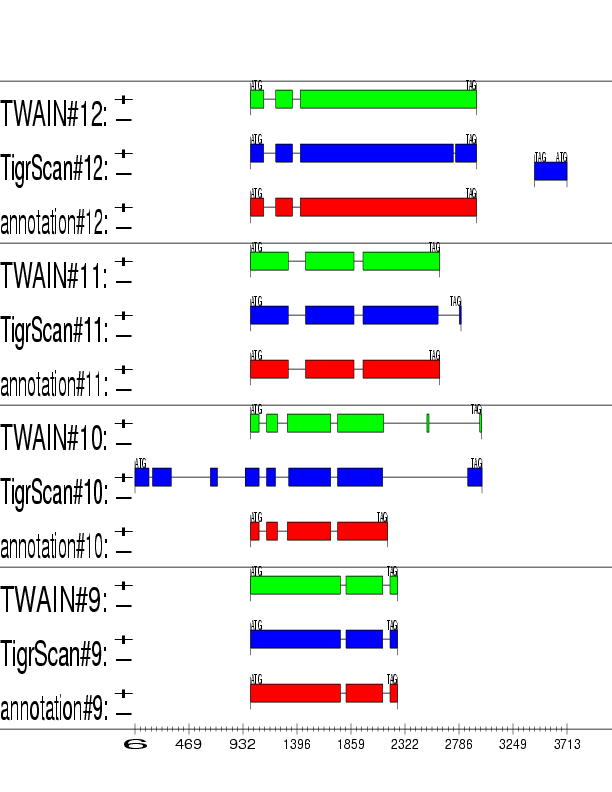
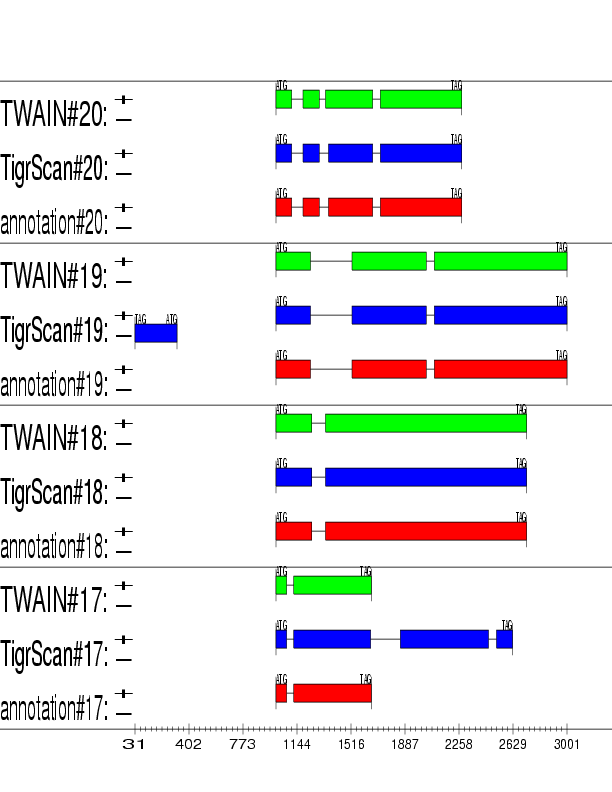
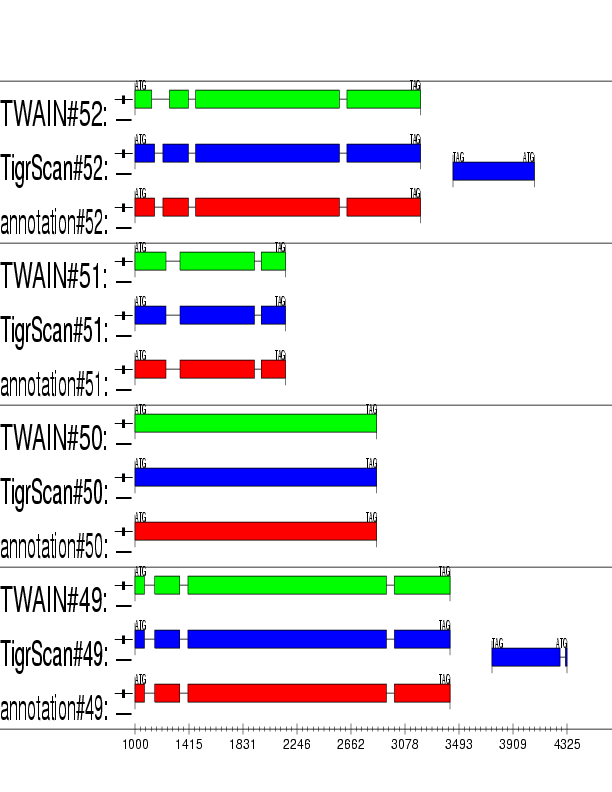
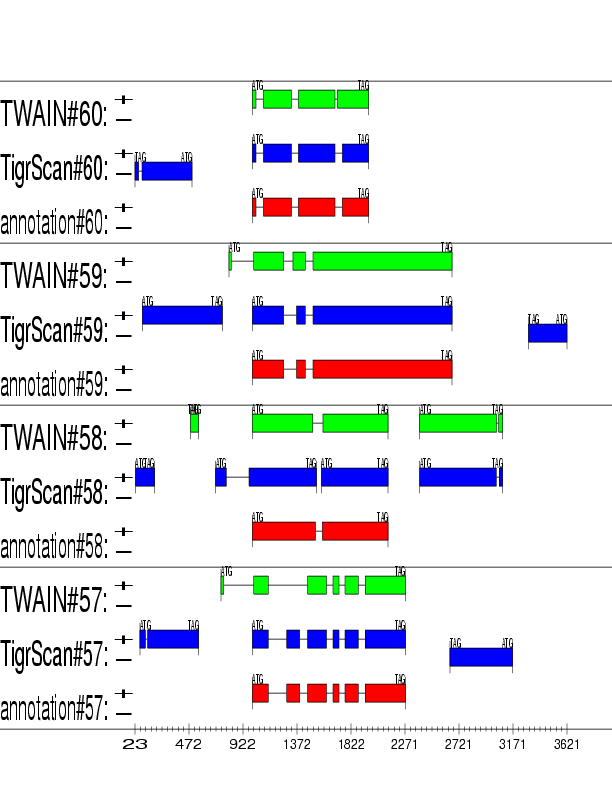
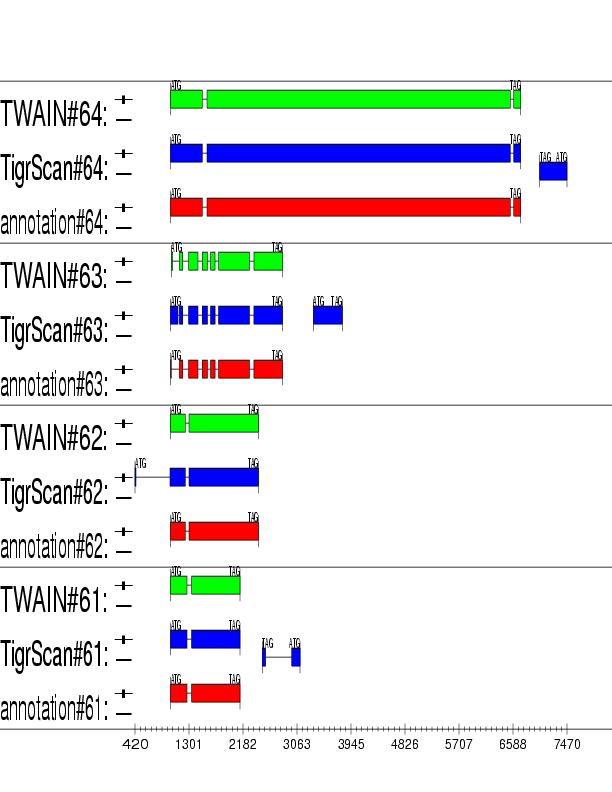
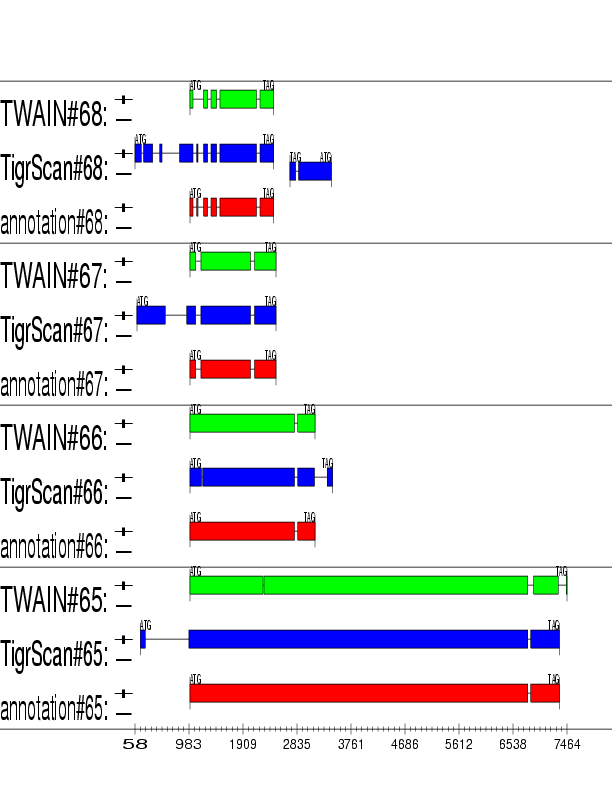
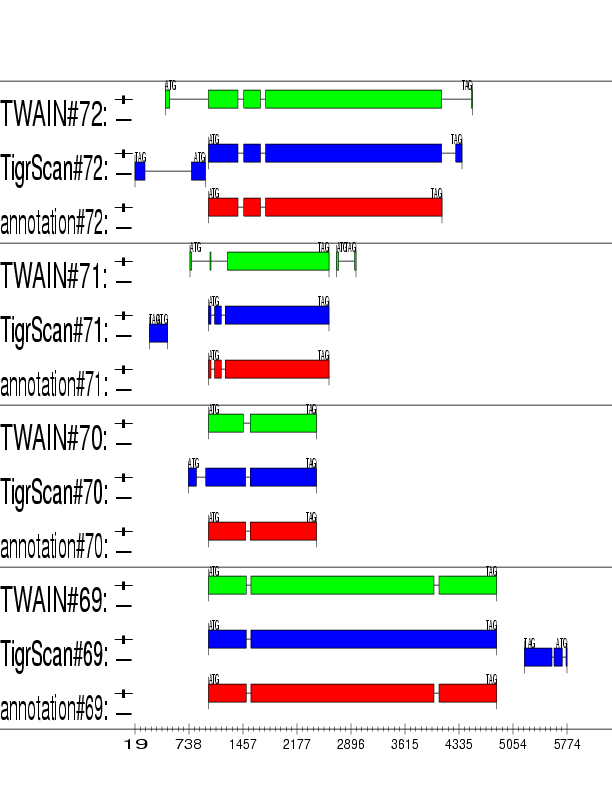
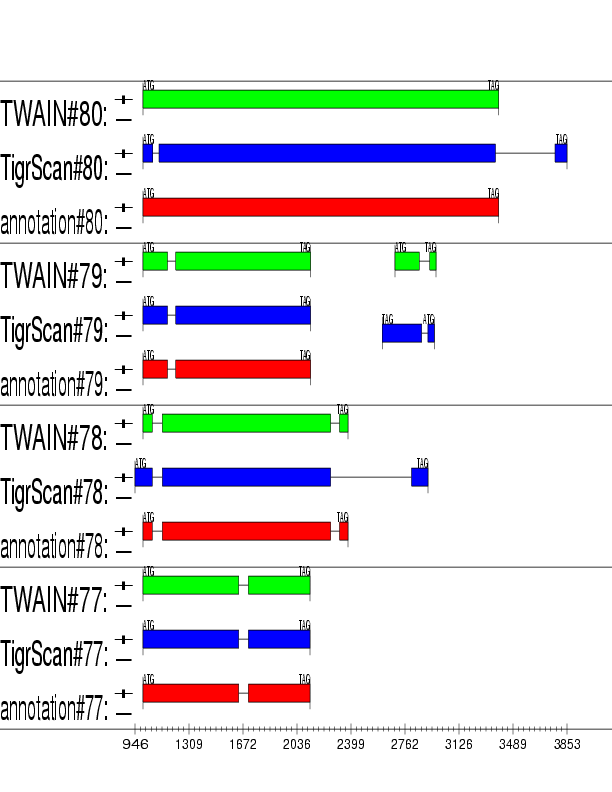
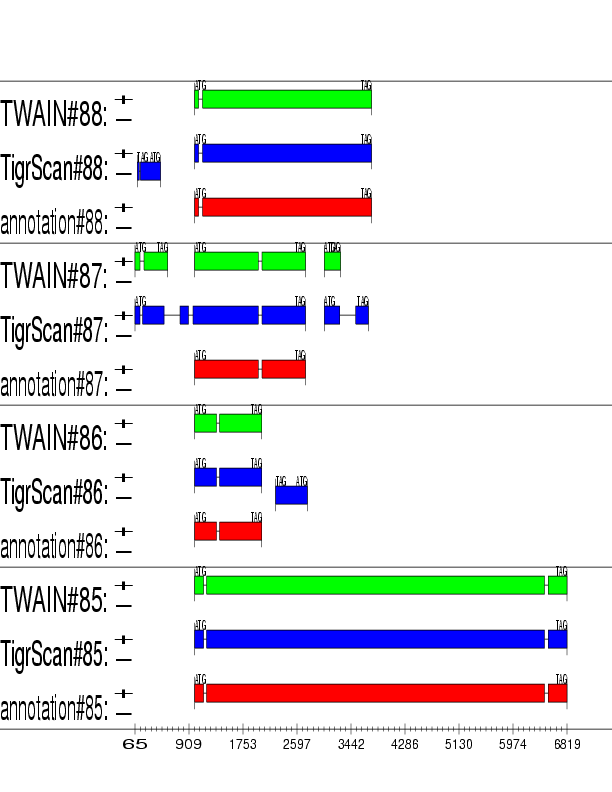
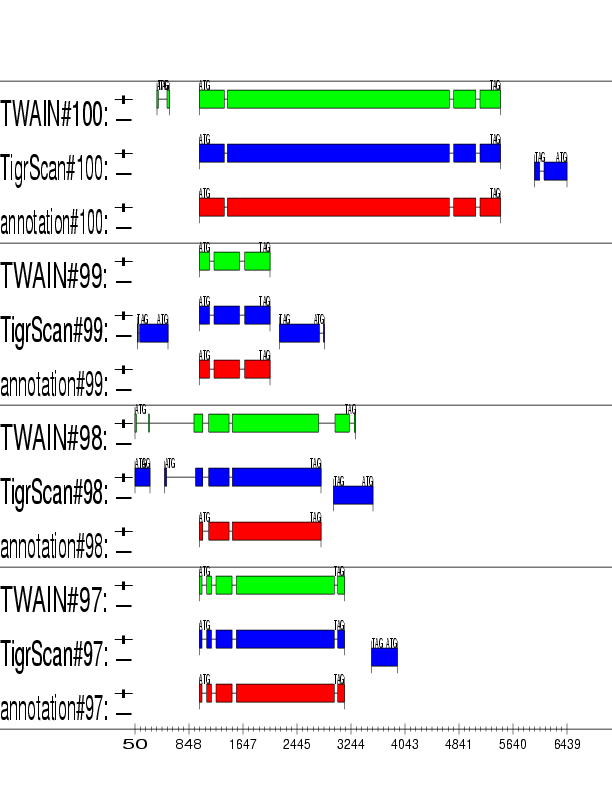
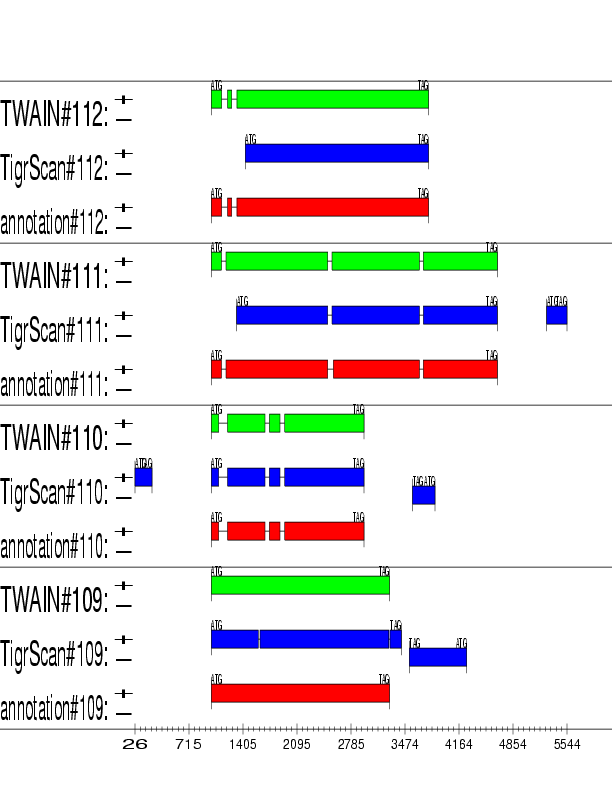
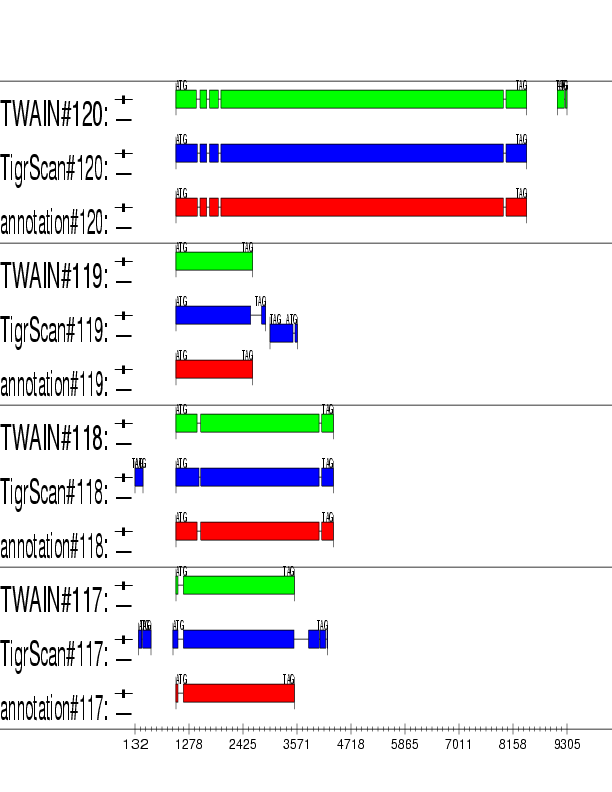
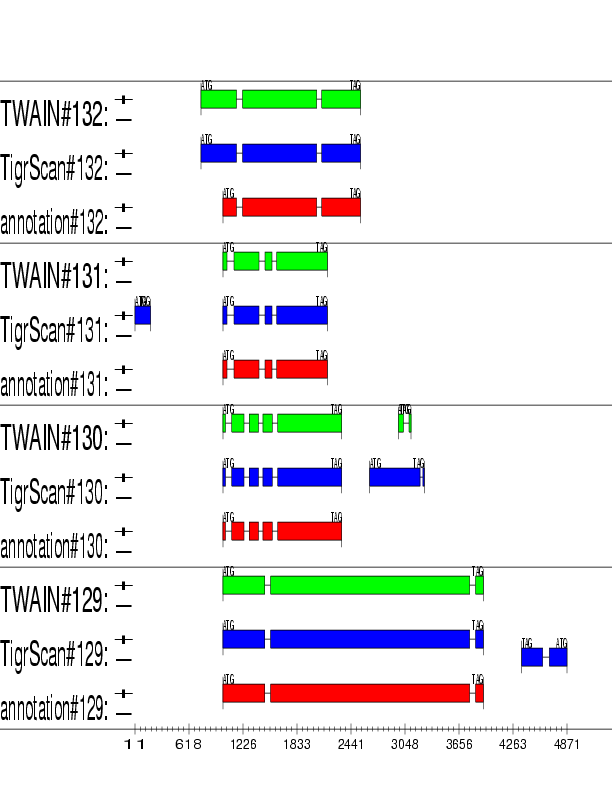
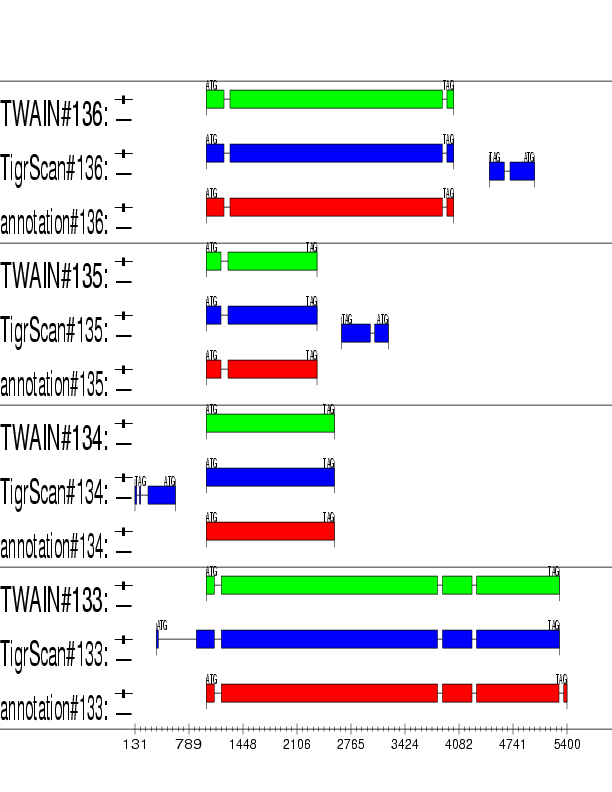
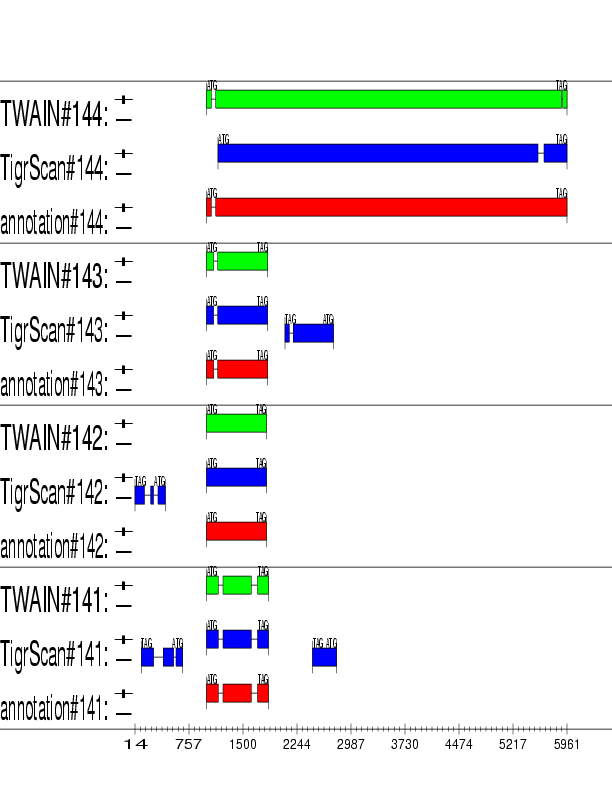
Example output:
Detailed Description
TWAIN consists of two components: (1) ROSE, the Region Of Synteny
Extractor, which identifies contiguous regions likely to contain
one or
more syntenic genes, and (2) OASIS, a generalized pair hidden Markov
model (GPHMM) for predicting genes in the regions identified by
ROSE. The system utilizes approximate alignments constructed by
the PROmer and NUCmer programs in the MUMmer package to
assess approximate alignment scores efficiently. More detailed
information on the architecture of this system will be made available
soon.
Slides from a talk at Computational Genomics 2004 are now
available.
Download
- MUMmer -
contains the PROmer and NUCmer programs used by ROSE
- ROSE - Region-Of-Synteny Extractor, a
preprocessor for the GPHMM
- OASIS - implements the GPHMM
- alignment - alignment package used
by OASIS to construct approximate alignments from MUMmer HSPs
- TigrScan
- used by OASIS to build a parse graph for each genome
- Documentation on using TWAIN
- Slides
from the talk at Computational
Genomics 2004
- Information on ROSE
- Sample files for a pair of fungal genomes:
- A. fumigatus models (*.cfg, *.model, *.iso,
*.top, *.distr, *.trans, train.sh)
- A. nidulans models (*.cfg, *.model, *.iso,
*.top, *.distr, *.trans, train.sh)
- training data
- fumigatus x nidulans syntenic pairs
References
Majoros
W.H., Pertea M., Salzberg S.L. (2005) Efficient implementation
of a Generalized Pair Hidden Markov Model for comparative gene finding.
Bioinformatics 21 1782-1788.
Aknowledgements
Development of TWAIN is supported by NIH under grant R01-LM06845.

|


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}